

业务痛点

行业监管要求
为贯彻落实《药品管理法》《疫苗管理法》有关规定,加强药品研制、生产、经营、使用活动的记录和数据管理,确保有关信息真实、准确、完整和可追溯。

编制问题
文件模板、版本不明确,编制人容易混乱;编制部门、编制人职责不明确;文件变更审批培训环节繁琐、不便捷、耗时多。

管理问题
文件(记录)管理工作量大,且逐年累积,造成混乱、错误、缺失等;文件总台账,收发记录等工作繁琐耗时、易出错

应用问题
不能快速查看检索文件文件内容;难以获取工作职责内的相关的文件和记录;借阅不便捷,且存在丢失的可能性
产品简介
睿展数据体系文件管理系统(DMS)遵循国家制药及医疗器械GMP规范要求,提供全生命周期的系统化流程化整体管理方案。系统满足生物医药行业生产管理和质量控制的基本要求,保证文件及记录管理的有关数据信息真实、准确、完整和可追溯。
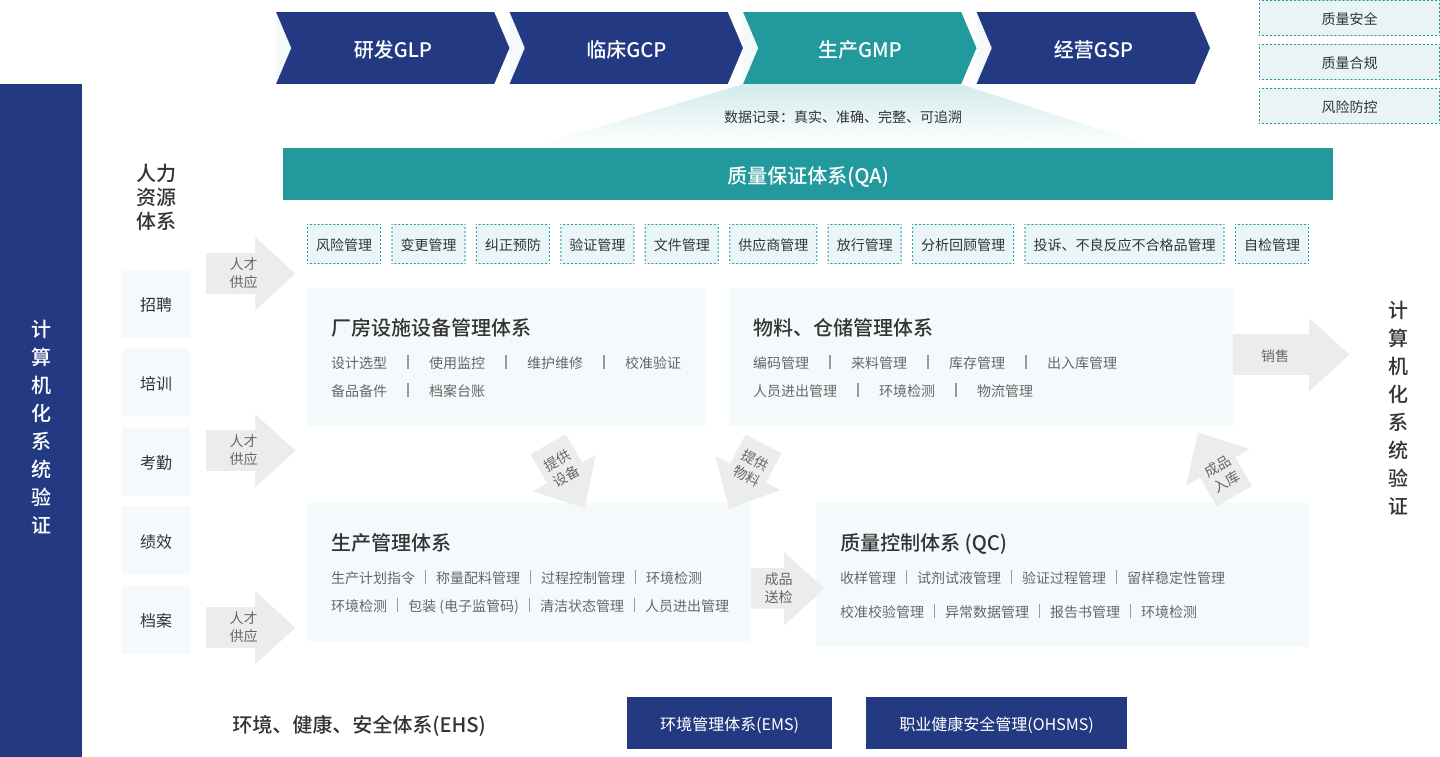
产品功能
通过构建DMS系统框架,让使用者对文件管理达到统一体系、集中管理、过程管控、持续改进的目的。
产品优势

灵活配置
文件变更及文件申请流程可依托企业管理机构特点、组织层级关系和管理思路差异,自行灵活定义配置。

降本增效
使用人员通过系统发起相关变更或应用流程,大量减少线下审核、批准、复制、分发,回收、归档的时间消耗;系统支持全文检索,文件查拢使用更方便快捷。

安全共享
系统满足计算机化系统所道循的数据安全法,系统内的文件、功能、业务、数据等均符合该法规要求,且可根据企业质量体系管理委求,实现权限控制下的电子数据的共享。

数据分析
体系文件管理人员通过系统统计分析实时点控体系文件变更,文件应用等,通过多维度的统计分析,即时的发现体系文件管理过程中的优势和欠缺等地方,是质量管理推进和完善的有力助手。

合法合规
基于国家药品/医疗器械生产质量管理规范、药品记录与数据管理要求、电子签名法以及美国,欧盟GMP等等法律法规的质量管理要求进行开发设计,满足GAMP5中对于的计算机化系统验证的要求,能完全实现数据审计追踪及数据完整性要求。

拓展延伸
文件作为GMP/1S0质量管理体系的基础和裁体,暨数提供体系文件与企业IT系统整体集成的能力,可与GXP业务框婴中及其他系统进行对接集成,比如:记录管理、偏差管理、变更管理、分析回顺管理、培训管理、生产管理、仓储物流管理、质控管理等等。
产品价值

体系文件结构化管理
通过IT化的方式,对体系文件进行结构化的管理,在体系文件内部、体系文件之间、体系文件与规范性引用文件、术语、报告、记录和工作职责之间建立建构化的关系,使体系文件实现数字化、应用对象化管理。

过程流程化管理
实现体系文件从变更(新增、修订、废止),审核,批准,培训,生效,发放,回收的全流程信息化管理闭环;并可根据企业的组织结构,管理方式对流程进行个性化定义。

体系文件应用与共享
为体系文件在企业各个工作场景中可以便捷的被关联和使用,系统能够提供多样、方便、快捷的使用方式和集成接入的通道。
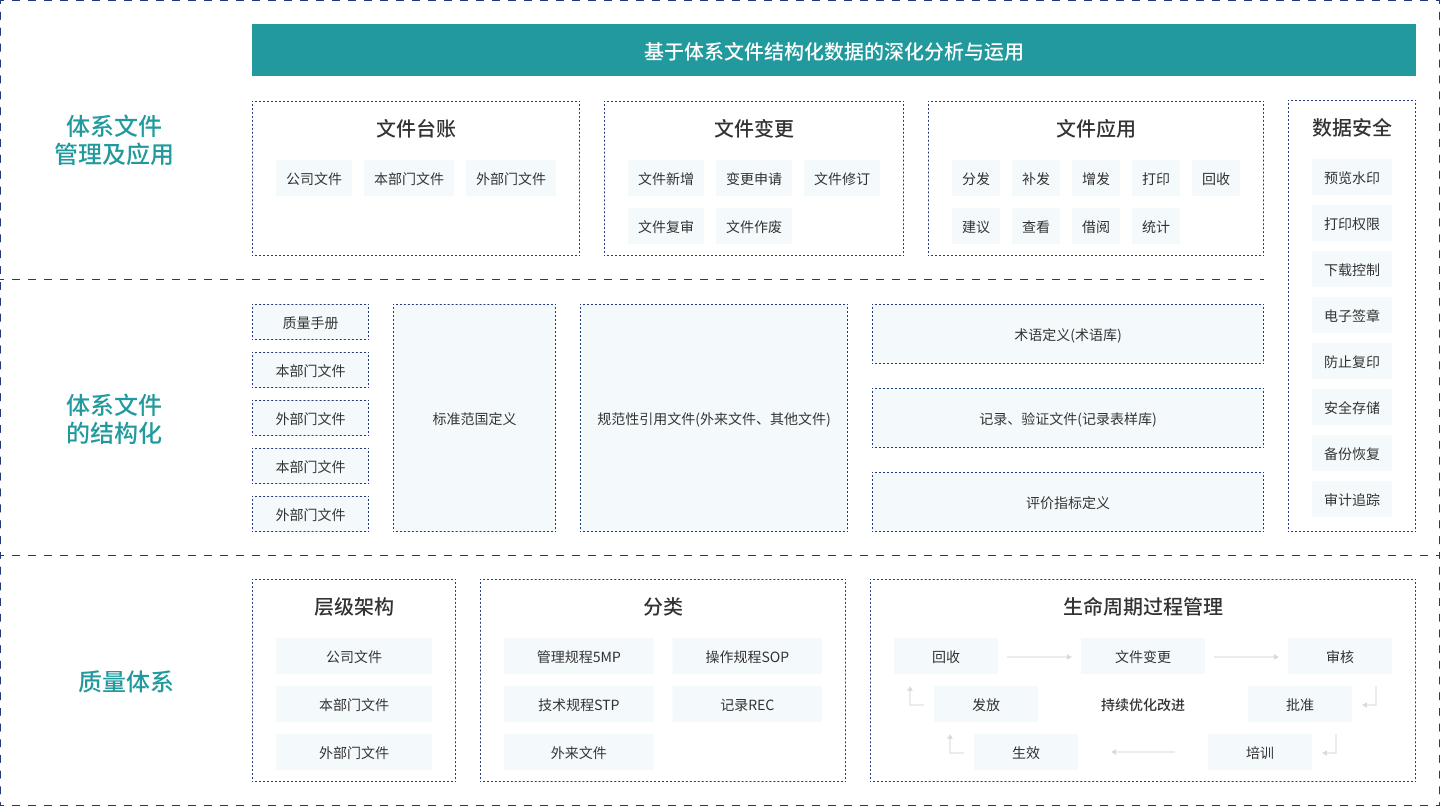
数据深化分析
通过文件变更数据、文件申请数据、文件应用数据的分析与运用,实现体系文件执行实时监控、关键指标实时预警,促进体系改善PDCA[计划(PLAN)、实施(DO)、检查(CHECK)、行动(ACTION)]循环。
 专精特新企业
专精特新企业 高新技术企业
高新技术企业 软件企业证书
软件企业证书 软件产品证书
软件产品证书 CMMI5认证
CMMI5认证
